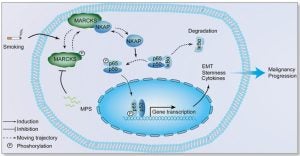
MARCKS cooperates with NKAP to activate NF-kB signaling in smoke-related lung cancer
Abstract
Rationale: Cigarette smoking is a major risk factor for lung cancer development and progression; however, the mechanism of how cigarette smoke activates signaling pathways in promoting cancer malignancy remains to be established. Herein, we aimed to determine the contribution of a signaling protein, myristoylated alanine-rich C kinase substrate (MARCKS), in smoke-mediated lung cancer.
Methods: We firstly examined the levels of phosphorylated M ARCKS (phospho-M ARCKS) in smoke-exposed human lung cancer cells and specimens as well as non-human primate airway epithelium. Next, the MARCKS-interactome and its gene networks were identified. We also used genetic and pharmacological approaches to verify the functionality and molecular mechanism of smoke-induced phospho-M ARCKS.
Results: We observed that MARCKS becomes activated in airway epithelium and lung cancer cells in response to cigarette smoke. Functional proteomics revealed MARCKS protein directly binds to NF-κB-activating protein (NKAP). Following MARCKS phosphorylation at ser159 and ser163, the MARCKS-NKAP interaction was inhibited, leading to the activation of NF-κB signaling. In a screen of two cohorts of lung cancer patients, we confirmed that phospho-MARCKS is positively correlated with phospho-NF-κB (phospho-p65), and poor survival. Surprisingly, smoke-induced phospho-MARCKS upregulated the expression of pro-inflammatory cytokines, epithelial–mesenchymal transition, and stem-like properties. Conversely, targeting of MARCKS phosphorylation with MPS peptide, a specific MARCKS phosphorylation inhibitor, suppressed smoke-mediated NF-κB signaling activity, pro-inflammatory cytokines expression, aggressiveness and stemness of lung cancer cells.
Conclusion: Our results suggest that phospho-MARCKS is a novel NF-kB activator in smoke-mediated lung cancer progression and provide a promising molecular model for developing new anticancer strategies.