Abstract
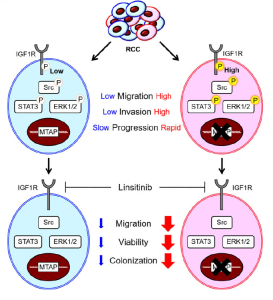
Renal cell carcinoma (RCC) has emerged as a metabolic disease characterized by dysregulated expression of metabolic enzymes. Patients with metastatic RCC have an unusually poor prognosis and near-universal resistance to all current therapies. To improve RCC treatment and the survival rate of patients with RCC, there is an urgent need to reveal the mechanisms by which metabolic reprogramming regulates aberrant signaling and oncogenic progression. Through an integrated analysis of RCC metabolic pathways, we showed that methylthioadenosine phosphorylase (MTAP) and its substrate methylthioadenosine (MTA) are dysregulated in aggressive RCC. A decrease in MTAP expression was observed in RCC tissues and correlated with higher tumor grade and shorter overall survival. Genetic manipulation of MTAP demonstrated that MTAP expression inhibits the epithelial-mesenchymal transition, invasion and migration of RCC cells. Interestingly, we found a decrease in the protein methylation level with a concomitant increase in tyrosine phosphorylation after MTAP knockout. A phospho-kinase array screen identified the type 1 insulin-like growth factor-1 receptor (IGF1R) as the candidate with the highest upregulation in tyrosine phosphorylation in response to MTAP loss. We further demonstrated that IGF1R phosphorylation acts upstream of Src and STAT3 signaling in MTAP-knockout RCC cells. IGF1R suppression by a selective inhibitor of IGF1R, linsitinib, impaired the cell migration and invasion capability of MTAP-deleted cells. Surprisingly, an increase in linsitinib-mediated cytotoxicity occurred in RCC cells with MTAP deficiency. Our data suggest that IGF1R signaling is a driver pathway that contributes to the aggressive nature of MTAP-deleted RCC.